Blogs & News
Avoiding GxP Non-Compliance with the Aridhia DRE
The pharmaceutical industry is highly regulated and subject to audits and compliance checks. The good manufacturing practices (GMP), good laboratory practices (GLP), good clinical practices (GCP), metrology, quality control (QC), and quality assurance (QA) are essential quality system frameworks and used in manufacturing and regulated environments, such as the drugs and pharmaceuticals, life science, and healthcare sectors. These quality frameworks collectively play a fundamental role in ensuring the safety, efficacy, and reliability of products and research within the life sciences. These standard practices, measurement, and quality system elements are intertwined, reinforcing one another to maintain the highest standards of safety, efficacy, and ethics throughout the life sciences [Rana 2024].
Adherence to these principles is an expectation of all pharmaceutical companies but the manner in which this is accomplished varies greatly and is often complicated with aspects of noncompliance along with various “workarounds” which ascribe to the “spirit of compliance.” There are differences Between GCP, GLP and GMP procedures and audits of course. Good Laboratory Practice (GLP) regulates the processes and conditions under which clinical and non-clinical research is conducted. GLP also governs how these research facilities should be maintained. Good Clinical Practice (GCP) guidelines are dictated by the International Conference on Harmonization (ICH). The ICH GCP governs the ethical and scientific quality of clinical trials. It covers things such as the study design, methodology, and data reporting related to clinical trials. Finally, Good Manufacturing Practice (GMP) regulates the design, monitoring, and control of manufacturing processes and facilities. GMP compliance, for example, ensures the identity, strength, quality, and purity of drug products.

The major differences between the three types of audits are related to the progressive stages needed to bring pharmaceutical, biologic, and medical device products to market. The main concern in the case of GCP is the health, safety, and rights of the study participant, as well as documentation that the product creates more benefits than harm. Public safety is particularly critical to inspectors. GLP does not involve human subjects, but nonclinical laboratory testing environments and materials. The main goal of GLP regulations is to ensure compliance in the conducting of nonclinical laboratory studies. It is a required step in obtaining premarket approval of regulated products. The main area of focus in GMP is the procedures of safe manufacturing, packaging, and processing of pharmaceutical products. Without the necessary clearances by the FDA, a drug cannot be brought to market. In any case, each of these well intended practices is subject to data generated by the company for the purpose of eventual regulatory review prior to registration and market access.
Data and Quality
Various data systems form the lifeblood of information and knowledge about a product in development, which is used for decision-making internally and the assembly of documentation (data, code and recommendations) that confirms the new chemical entity’s safety and efficacy in the target population along with all the steps to robustly produce and manufacture the drug product [Oronsky 2023]. These include various laboratory information management systems (LIMS), Clinical data management systems (CDMs) and document management systems (DMS). Quality has become increasingly important to companies as stakeholder groups, interest groups and members of the public require that their concerns about certain aspects of companies’ operations are taken into account by managers of the companies. Unforeseen consequences raise people’s and authorities’ awareness. GLP principles set out standards for conducting nonclinical research. GLP principles are stipulated in law, but are not typically well known. As drug development and research in general are highly data driven, these three governing principles (GLP, GCP and GMP) are affected and impacted by data fundamental to decision-making as well as the evidence that drug candidates submitted for regulatory approval are safe and efficacious.
The flow of data within an organization is influenced by the functional groups that generate, manage and provide data for regulatory submission (frequently different groups with different understanding about data quality, data integrity and data standards. The flow of data is defined by the underlying architecture that stores, transforms and communicates data in some form or another to both internal and external stakeholders. Depending on the requirements for each one of these solutions, the manner in which these systems interact with each other may not be optimal or efficient. While there may be some oversight from an IT perspective, the final say is typically decided by the functional group that defines the requirements and pays the bill. Likewise, while GLP, GCP and GMP is intended by the company and the supporting functional groups, their choices with respect to these core systems may make these principles vulnerable.
GLP, GCP, and GMP
GLP, GCP, and GMP regulations pertaining to testing serve different purposes as previously mentioned. With respect to data, GLPs are designed to protect scientific data integrity, and to provide the EPA or FDA with a clear and auditable record of open-ended research studies. The GCPs are intended to be an ethical and scientific quality standard for designing, conducting, recording, and reporting studies that involve the participation of human subjects. Compliance with this standard provides public assurance that the rights, safety, and wellbeing of clinical study subjects are protected, consistent with the principles that have their origin in the Declaration of Helsinki, and that the clinical data are credible. Finally, the GMPs are intended to demonstrate to the FDA whether or not individual batches of a regulated product are manufactured according to pre-defined manufacturing criteria. Hence, while these principles serve different purpose and impact different stages of development and distinct aspects of development, there is synergy between GMP, GLP, GCP, metrology, quality control (QC), and quality assurance (QA). Likewise, data is the backbone upon which these principles are demonstrated and judged. Figure 1 provides a schematic overview between these principles demonstrating synergy and the connectivity to core data systems that capture, manage, and exploit the underlying data.

Figure 1. Relationship between GLP, GCP, and GMP regulations, sponsors’ activities and core data systems used to manage guideline and data-related procedures.
The seamless integration of these principles across GMP, GLP, GCP, metrology, QC, and QA ultimately serves to safeguard patients and uphold the integrity of research and product development in the healthcare and pharmaceutical sectors. Likewise, the core data systems also provide physical proof of the data and research integrity and must operate in a validated, secure manner in compliance with the code of federal regulations. Figure 2 provides an overview of the key challenges encountered in R&D and commercial operations of a pharmaceutical sponsor related to synergy (or lack of) between GMP, GLP, GCP, metrology, QC, and QA [Rana 2023].
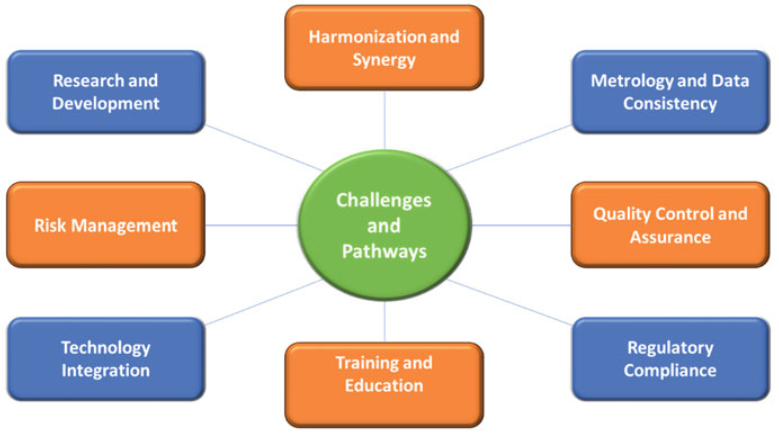
Figure 2. Challenges and pathways for synergy between GMP, GLP, GCP, metrology, QC, and QA [adopted from Rana et al 2023].
What is not defined or apparent in Figure 2 are the vast number of shortcuts, workarounds, or other measures a sponsor utilizes to manage performance expectations while simultaneously creating compliance and audit vulnerabilities. The nature of workarounds is varied and can include any number of data practices which are problematic. A workaround has been defined as “a goal-driven adaptation, improvisation, or other change to one or more aspects of an existing work system in order to overcome, bypass, or minimize the impact of obstacles, exceptions, anomalies, mishaps, established practices, management expectations, or structural constraints that are perceived as preventing that work system or its participants from achieving a desired level of efficiency, effectiveness, or other organizational or personal goals. [Alter 2014].” This definition, however, is very broad, tying together quick fixes of technology mishaps, shadow IT systems, and deviations from the required course of action in business processes.
Having had the responsibility to consolidate compute services for modelling and simulation groups in the past, I’ve been struck by the allegiance to antiquated legacy systems, particularly those that are siloed, unsecure, and difficult to use. The “if it ain’t broke don’t fix it cliché” should be ringing in your ears now. The underlying reasons that these situations persist can include everything from loyalty to the internal specialist who created and still maintains the solution (hardware, software or combination), perceived cost efficiency or at least neutrality, autonomy for maintenance by the functional group and the lack of necessity to learn something new. Another common workaround category is the mechanism by which various functional groups access and transfer data. There are still occasions where email attachments, USB drives and other rudimentary means are utilized as part of workarounds to get data from CROs, LIMS systems and other operational data stores. For many of these workarounds the motivation is the limitations of either old hardware or outdated data solutions. The combination of issues makes compliance with the 3 principles (GLP, GCP and GCP) and event internal SOPs problematic.
Control and Standard Operating Procedures
Prolonged noncompliance creates vulnerabilities as previously discussed and also a lack of control. Elements of management control systems include strategic planning, budgeting, resource allocation, performance measurement, evaluation, reward, responsibility centre allocation and transfer pricing [Anthony et al 2003]. The overall purpose of a management system is to help a company to implement its strategies. When a company decides to implement a quality system as a part of its management system, this can enhance its performance in many ways. Improved quality can affect profitability in many ways. A consequence can be the lack of integration oversight, particularly with related or complimentary functional groups (e.g., Biostatistics / statistical programming and Clinical Pharmacology / Modelling and Simulation). More problematic are the groups that behave in a more siloed manner because of their deliverables, particularly those that have different functional alignment / reporting relationships (e.g., Clinical Operations).

Standard Operating Procedures (SOP) attempt to document the way that an operator should perform a given operation. The SOP is one of many process documents which is needed for consistent operation of a given process, with other documents involving process flow charts, material specifications, and so forth. Clearly the aforementioned workarounds are outside SOP specifications making noncompliance with internal SOPs a problem and a risk with respect to any regulatory audits. While some sponsors engage in mock audits these tend to concentrate on individual projects and project data and the solution is a somewhat staged preparation for a specific audit as opposed to an internal risk assessment and compliance check.
The Aridhia DRE provides a rich metadata catalogue with advanced searching and support for FAIR data principles. From a compliance standpoint the configurable and orchestrated data governance automatically connects metadata searches with data requests and delivery and allows connectivity with legacy systems which may be problematic in a siloed existence (see Figure 3 below). Private, airlocked, and data-agnostic collaboration workspaces operate in a scalable and audited regulatory-grade environment, reducing the necessity of workarounds. The DRE solution is highly customizable and has been pressure tested in both single institution environments (GOSH/UCL, Royal Marsden Hospital, etc.) and collaborative research and data sharing ecosystems (ICODA, C-PATH / RDCA-DAP, AD Data Initiative, etc).
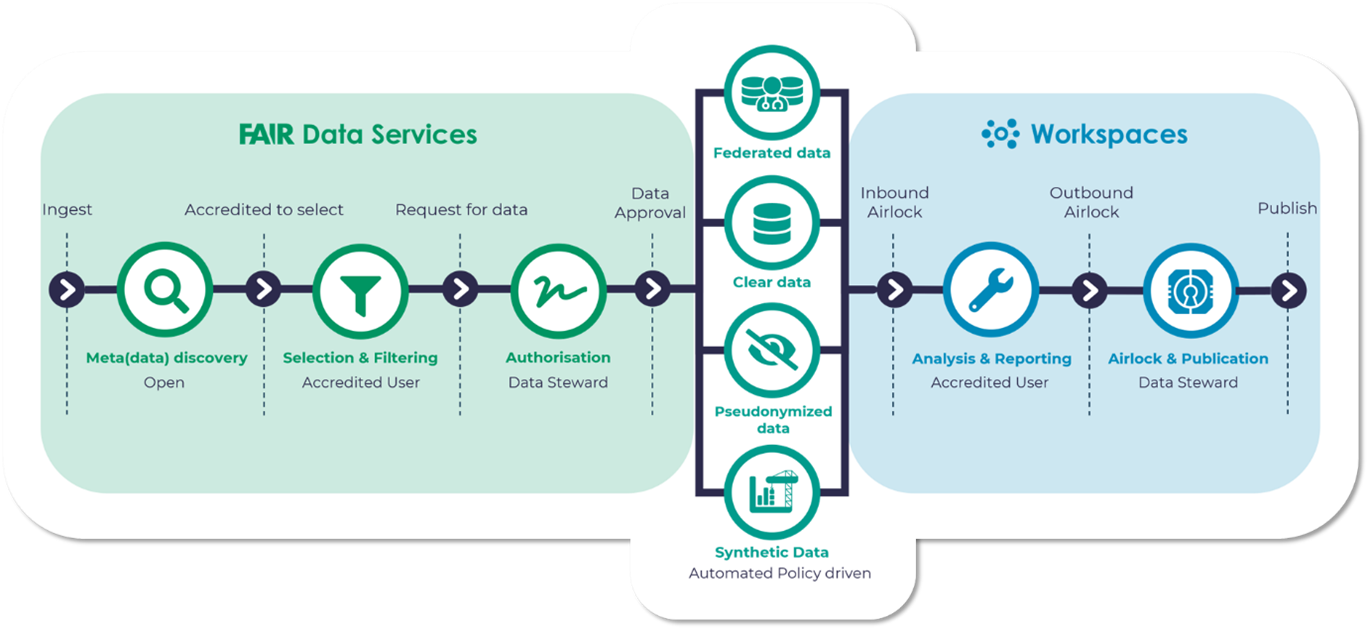
Figure 3. Overview of DRE FAIR and Workspaces services.
Additional detailed examples can be found on the Aridhia website along with published examples of the solution implementations. Feel free to contact me or any member of our staff for a tailored demo on how the DRE can obviate the need for workarounds, ensure compliance with SOPs and ultimately remove risks associated with GLP, GCP and GMP noncompliance.
References
- • Rana, B.K., Yadav, D., Yadav, S. (2024). Strategic Association for Quality Excellence: Coordination Between GMP, GLP, GCP, Metrology, QC, and QA. In: Bhatnagar, A., Yadav, S., Achanta, V., Harmes-Liedtke, U., Rab, S. (eds) Handbook of Quality System, Accreditation and Conformity Assessment. Springer, Singapore.
- • Oronsky B, Burbano E, Stirn M, Brechlin J, Abrouk N, Caroen S, Coyle A, Williams J, Cabrales P, Reid TR. Data Management 101 for drug developers: A peek behind the curtain. Clin Transl Sci. 2023 Sep;16(9):1497-1509
- • Alter S. Theory of workarounds. Commun. Assoc. Inf. Syst. 2014;34(1):1041–1066
- • Barrett JS, editor, Fundamentals of Drug Development, Wiley; 1st edition (September 7, 2022), ISBN-10: 1119691699, ISBN-13: 978-1119691693 (512 pages).
- • Dixon JR Jr. The International Conference on Harmonization Good Clinical Practice guideline. Qual Assur. 1998 Apr-Jun;6(2):65-74
- • International Ethical Guidelines for Health-related Research Involving Humans, Fourth Edition. Geneva. Council for International Organizations of Medical Sciences (CIOMS); 2016
- • Goodyear MD, Krleza-Jeric K, Lemmens T. The Declaration of Helsinki. BMJ. 2007 Sep 29;335(7621):624-5
- • Outmazgin N, Soffer P, Hadar I. Workarounds in Business Processes: A Goal-Based Analysis. Advanced Information Systems Engineering. 2020 May 9;12127:368–83